Alkaptonuria
Published: 18 Jun 2025
ICD9: 270.2 ICD10: E70 ICD11: 5C50.10
Alkaptonuria (AKU), also known as "black urine disease," is a rare inherited genetic disorder characterized by the body's inability to properly process the amino acids tyrosine and phenylalanine.
This deficiency stems from a defect in the enzyme homogentisate 1,2-dioxygenase (HGD).
Here's a breakdown of key aspects of Alkaptonuria:
![]() Cause: A mutation in the *HGD* gene, which provides instructions for making the homogentisate 1,2-dioxygenase enzyme. This enzyme is crucial for breaking down homogentisic acid (HGA), a byproduct of tyrosine and phenylalanine metabolism.
Cause: A mutation in the *HGD* gene, which provides instructions for making the homogentisate 1,2-dioxygenase enzyme. This enzyme is crucial for breaking down homogentisic acid (HGA), a byproduct of tyrosine and phenylalanine metabolism.![]() What happens: Without a functional HGD enzyme, homogentisic acid accumulates in the body. This excess HGA deposits in various tissues, leading to the characteristic symptoms of AKU.
What happens: Without a functional HGD enzyme, homogentisic acid accumulates in the body. This excess HGA deposits in various tissues, leading to the characteristic symptoms of AKU.![]() Symptoms: The most common symptoms develop over time due to the buildup of HGA:
Symptoms: The most common symptoms develop over time due to the buildup of HGA:![]()

![]() Darkening of Urine: This is often the first noticeable sign, even in infancy. Urine turns dark when exposed to air.
Darkening of Urine: This is often the first noticeable sign, even in infancy. Urine turns dark when exposed to air.![]()
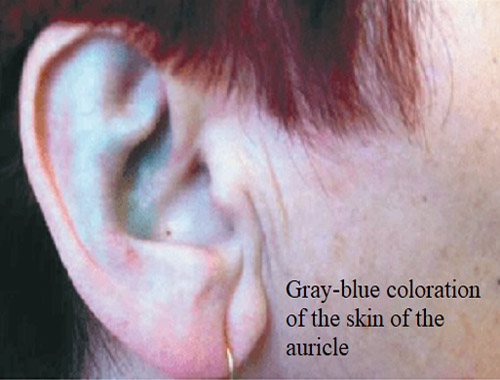
![]() Ochronosis: Bluish-black pigmentation of cartilage and other connective tissues, particularly noticeable in the ears, nose, and sclera (whites of the eyes).
Ochronosis: Bluish-black pigmentation of cartilage and other connective tissues, particularly noticeable in the ears, nose, and sclera (whites of the eyes).![]()
![]() Arthritis: HGA deposits in joints, causing pain, stiffness, and eventually osteoarthritis, often starting in the spine and large joints (hips, knees, shoulders). This is a major source of morbidity in AKU.
Arthritis: HGA deposits in joints, causing pain, stiffness, and eventually osteoarthritis, often starting in the spine and large joints (hips, knees, shoulders). This is a major source of morbidity in AKU.![]()
![]() Kidney and Prostate Stones: HGA can contribute to the formation of kidney and prostate stones.
Kidney and Prostate Stones: HGA can contribute to the formation of kidney and prostate stones.![]()
![]() Cardiac Problems: In some cases, HGA can affect the heart valves.
Cardiac Problems: In some cases, HGA can affect the heart valves.![]() Inheritance: It's an autosomal recessive disorder. This means that a person must inherit two copies of the mutated gene (one from each parent) to develop AKU. Individuals with only one copy of the mutated gene are carriers and usually don't show symptoms.
Inheritance: It's an autosomal recessive disorder. This means that a person must inherit two copies of the mutated gene (one from each parent) to develop AKU. Individuals with only one copy of the mutated gene are carriers and usually don't show symptoms.![]() Diagnosis:
Diagnosis:![]()
![]() Urine Test: Elevated levels of homogentisic acid in urine are a key diagnostic marker.
Urine Test: Elevated levels of homogentisic acid in urine are a key diagnostic marker.![]()
![]() Genetic Testing: Can confirm the diagnosis by identifying mutations in the *HGD* gene.
Genetic Testing: Can confirm the diagnosis by identifying mutations in the *HGD* gene.![]()
![]() Physical Examination: Examination of the ears and eyes can reveal ochronosis.
Physical Examination: Examination of the ears and eyes can reveal ochronosis.![]() Treatment:
Treatment:![]()
![]() Nitisinone: This medication inhibits an enzyme earlier in the tyrosine/phenylalanine pathway, reducing the production of homogentisic acid. It's currently the only approved medication for AKU and helps to slow the progression of the disease.
Nitisinone: This medication inhibits an enzyme earlier in the tyrosine/phenylalanine pathway, reducing the production of homogentisic acid. It's currently the only approved medication for AKU and helps to slow the progression of the disease.![]()
![]() Dietary Restrictions: Limiting protein intake (particularly tyrosine and phenylalanine) *may* help, but it is difficult to achieve and its long-term efficacy is debated. It is generally recommended to maintain a normal, healthy diet.
Dietary Restrictions: Limiting protein intake (particularly tyrosine and phenylalanine) *may* help, but it is difficult to achieve and its long-term efficacy is debated. It is generally recommended to maintain a normal, healthy diet.![]()
![]() Pain Management: Analgesics and anti-inflammatory medications can help manage joint pain.
Pain Management: Analgesics and anti-inflammatory medications can help manage joint pain.![]()
![]() Physical Therapy: Can help maintain joint mobility and strength.
Physical Therapy: Can help maintain joint mobility and strength.![]()
![]() Joint Replacement: May be necessary in severe cases of arthritis.
Joint Replacement: May be necessary in severe cases of arthritis.![]()
![]() Monitoring: Regular monitoring for kidney stones, prostate stones, and cardiac issues is important.
Monitoring: Regular monitoring for kidney stones, prostate stones, and cardiac issues is important.![]() Prognosis: Alkaptonuria is a chronic condition, but with treatment and management, individuals can often live relatively normal lives. Early diagnosis and treatment with nitisinone are crucial to slow the progression of the disease and prevent long-term complications.
Prognosis: Alkaptonuria is a chronic condition, but with treatment and management, individuals can often live relatively normal lives. Early diagnosis and treatment with nitisinone are crucial to slow the progression of the disease and prevent long-term complications.
In summary, Alkaptonuria is a rare genetic disorder characterized by the accumulation of homogentisic acid in the body, leading to dark urine, ochronosis, arthritis, and other complications. Nitisinone is the primary treatment, aiming to reduce HGA production and slow disease progression. Early diagnosis and ongoing management are essential for improving the quality of life for individuals with AKU.