Amyotrophic lateral sclerosis (ALS)
Published: 18 Jun 2025
ICD9: 335.20 ICD10: G12.21 ICD11: 8B60.0
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, is a progressive neurodegenerative disease that affects nerve cells in the brain and spinal cord.
It causes muscle weakness, paralysis, and ultimately, respiratory failure. Here's a more detailed breakdown:
Key Aspects of ALS:
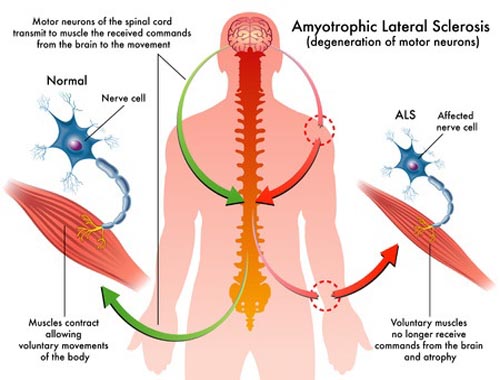
![]() Neurodegenerative: The disease involves the gradual destruction of neurons (nerve cells) responsible for controlling voluntary muscle movement. These are specifically motor neurons.
Neurodegenerative: The disease involves the gradual destruction of neurons (nerve cells) responsible for controlling voluntary muscle movement. These are specifically motor neurons.![]() Motor Neurons: ALS primarily affects two types of motor neurons:
Motor Neurons: ALS primarily affects two types of motor neurons:![]()

![]() Upper Motor Neurons: Located in the brain, these neurons send messages to the lower motor neurons. Damage to these causes stiffness (spasticity) and exaggerated reflexes.
Upper Motor Neurons: Located in the brain, these neurons send messages to the lower motor neurons. Damage to these causes stiffness (spasticity) and exaggerated reflexes.![]()
![]() Lower Motor Neurons: Located in the spinal cord and brainstem, these neurons transmit signals to muscles. Damage to these causes muscle weakness, twitching (fasciculations), and muscle atrophy (wasting).
Lower Motor Neurons: Located in the spinal cord and brainstem, these neurons transmit signals to muscles. Damage to these causes muscle weakness, twitching (fasciculations), and muscle atrophy (wasting).![]() Progressive: ALS gets worse over time. Symptoms start gradually and progressively worsen until muscles are paralyzed. The rate of progression varies significantly from person to person.
Progressive: ALS gets worse over time. Symptoms start gradually and progressively worsen until muscles are paralyzed. The rate of progression varies significantly from person to person.![]() Amyotrophic: This term comes from Greek and refers to the muscle atrophy (wasting) that occurs because the muscles aren't receiving signals from the nerves. "A" means no/without, "myo" refers to muscle, and "trophic" means nourishment.
Amyotrophic: This term comes from Greek and refers to the muscle atrophy (wasting) that occurs because the muscles aren't receiving signals from the nerves. "A" means no/without, "myo" refers to muscle, and "trophic" means nourishment.![]() Lateral sclerosis: This refers to the hardening (sclerosis) of the lateral (side) portions of the spinal cord, which occurs as the motor neurons degenerate and are replaced by scar tissue.
Lateral sclerosis: This refers to the hardening (sclerosis) of the lateral (side) portions of the spinal cord, which occurs as the motor neurons degenerate and are replaced by scar tissue.
Symptoms:
Symptoms vary depending on which motor neurons are affected first, but common symptoms include:![]() Muscle Weakness: Often the first symptom, starting in the hands, feet, arms, or legs. Can also affect muscles involved in speech and swallowing.
Muscle Weakness: Often the first symptom, starting in the hands, feet, arms, or legs. Can also affect muscles involved in speech and swallowing.![]() Muscle Twitching and Cramps: Fasciculations (small muscle twitches) are common.
Muscle Twitching and Cramps: Fasciculations (small muscle twitches) are common.![]() Slurred Speech and Difficulty Swallowing (Dysarthria and Dysphagia): These symptoms occur when the muscles controlling speech and swallowing are affected.
Slurred Speech and Difficulty Swallowing (Dysarthria and Dysphagia): These symptoms occur when the muscles controlling speech and swallowing are affected.![]() Difficulty Breathing: As the muscles controlling breathing weaken, shortness of breath and fatigue develop. Eventually, respiratory failure occurs.
Difficulty Breathing: As the muscles controlling breathing weaken, shortness of breath and fatigue develop. Eventually, respiratory failure occurs.![]() Stiffness (Spasticity): Muscles may become stiff and tight, limiting movement.
Stiffness (Spasticity): Muscles may become stiff and tight, limiting movement.![]() Changes in Cognition and Behavior: While ALS primarily affects motor function, some individuals may experience cognitive and behavioral changes, including problems with memory, decision-making, or personality. Frontotemporal dementia (FTD) can be associated with ALS in some cases.
Changes in Cognition and Behavior: While ALS primarily affects motor function, some individuals may experience cognitive and behavioral changes, including problems with memory, decision-making, or personality. Frontotemporal dementia (FTD) can be associated with ALS in some cases.
Causes:
The exact cause of ALS is not fully understood in most cases.![]() Genetic Factors: In a small percentage of cases (around 5-10%), ALS is inherited (familial ALS). Several genes have been identified that are associated with ALS.
Genetic Factors: In a small percentage of cases (around 5-10%), ALS is inherited (familial ALS). Several genes have been identified that are associated with ALS.![]() Sporadic ALS: Most cases of ALS are sporadic, meaning they occur randomly without a known family history. Researchers are investigating various factors that may contribute to sporadic ALS, including:
Sporadic ALS: Most cases of ALS are sporadic, meaning they occur randomly without a known family history. Researchers are investigating various factors that may contribute to sporadic ALS, including:![]()
![]() Glutamate Excitotoxicity: Excessive glutamate, a neurotransmitter, can damage motor neurons.
Glutamate Excitotoxicity: Excessive glutamate, a neurotransmitter, can damage motor neurons.![]()
![]() Protein Misfolding and Aggregation: Accumulation of misfolded proteins within motor neurons.
Protein Misfolding and Aggregation: Accumulation of misfolded proteins within motor neurons.![]()
![]() Oxidative Stress: Damage from free radicals.
Oxidative Stress: Damage from free radicals.![]()
![]() Mitochondrial Dysfunction: Problems with the energy-producing parts of cells.
Mitochondrial Dysfunction: Problems with the energy-producing parts of cells.![]()
![]() Environmental Factors: Some studies suggest possible links to environmental toxins, heavy metals, or other exposures, but more research is needed.
Environmental Factors: Some studies suggest possible links to environmental toxins, heavy metals, or other exposures, but more research is needed.
Diagnosis:
Diagnosing ALS can be challenging because there is no single definitive test. Doctors typically use a combination of:![]() Neurological Examination: To assess muscle strength, reflexes, coordination, and sensation.
Neurological Examination: To assess muscle strength, reflexes, coordination, and sensation.![]() Electromyography (EMG): To measure electrical activity in muscles and detect nerve damage.
Electromyography (EMG): To measure electrical activity in muscles and detect nerve damage.![]() Nerve Conduction Studies (NCS): To measure the speed of electrical signals traveling along nerves.
Nerve Conduction Studies (NCS): To measure the speed of electrical signals traveling along nerves.![]() Magnetic Resonance Imaging (MRI): Of the brain and spinal cord to rule out other conditions.
Magnetic Resonance Imaging (MRI): Of the brain and spinal cord to rule out other conditions.![]() Blood and Urine Tests: To rule out other possible causes of the symptoms.
Blood and Urine Tests: To rule out other possible causes of the symptoms.![]() Lumbar Puncture (Spinal Tap): To analyze cerebrospinal fluid.
Lumbar Puncture (Spinal Tap): To analyze cerebrospinal fluid.
Treatment:
There is no cure for ALS. Treatment focuses on managing symptoms, slowing disease progression, and improving quality of life. This typically involves:![]() Medications:
Medications:![]()
![]() Riluzole: Can modestly slow the progression of ALS by reducing glutamate levels.
Riluzole: Can modestly slow the progression of ALS by reducing glutamate levels.![]()
![]() Edaravone: An antioxidant that may slow the decline in physical function in some individuals.
Edaravone: An antioxidant that may slow the decline in physical function in some individuals.![]()
![]() Tofersen (Qalsody): For individuals with ALS caused by a mutation in the SOD1 gene.
Tofersen (Qalsody): For individuals with ALS caused by a mutation in the SOD1 gene.![]() Therapy:
Therapy:![]()
![]() Physical Therapy: To maintain muscle strength, flexibility, and range of motion.
Physical Therapy: To maintain muscle strength, flexibility, and range of motion.![]()
![]() Occupational Therapy: To help with daily activities and adapt to changing abilities.
Occupational Therapy: To help with daily activities and adapt to changing abilities.![]()
![]() Speech Therapy: To improve communication and swallowing.
Speech Therapy: To improve communication and swallowing.![]()
![]() Respiratory Therapy: To manage breathing difficulties.
Respiratory Therapy: To manage breathing difficulties.![]() Nutritional Support: To maintain adequate nutrition and prevent weight loss.
Nutritional Support: To maintain adequate nutrition and prevent weight loss.![]() Assistive Devices: Including wheelchairs, walkers, communication devices, and breathing support equipment.
Assistive Devices: Including wheelchairs, walkers, communication devices, and breathing support equipment.![]() Palliative Care: To provide comfort and support to patients and their families.
Palliative Care: To provide comfort and support to patients and their families.
Prognosis:
The prognosis for ALS varies. Most people with ALS live for 2-5 years after diagnosis, but some live much longer. Factors that can influence prognosis include age at diagnosis, the type of ALS (e.g., bulbar vs. limb onset), and the rate of disease progression.
In summary, ALS is a devastating disease that progressively destroys motor neurons, leading to muscle weakness, paralysis, and ultimately death. While there is no cure, treatments are available to manage symptoms and improve quality of life.