Bruton (X-linked) agammaglobulinemia
Published: 18 Jun 2025
ICD9: 279.04 ICD10: D80.0 ICD11: 4A01.00
Bruton (X-linked) agammaglobulinemia (XLA) is a rare genetic disorder that affects the immune system.
It primarily affects males and is characterized by a near or complete absence of mature B cells and very low levels of antibodies (immunoglobulins) in the blood.
Here's a breakdown of key aspects:
![]() Genetic Basis:
Genetic Basis:![]()

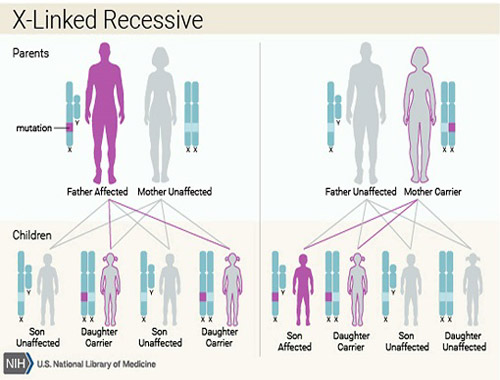
![]() XLA is caused by mutations in the *BTK* (Bruton's tyrosine kinase) gene located on the X chromosome.
XLA is caused by mutations in the *BTK* (Bruton's tyrosine kinase) gene located on the X chromosome.![]()
![]() BTK is crucial for the development and maturation of B cells. A faulty BTK gene means B cells can't mature properly, leading to their absence in the bloodstream.
BTK is crucial for the development and maturation of B cells. A faulty BTK gene means B cells can't mature properly, leading to their absence in the bloodstream.![]()
![]() Since males have one X chromosome (XY), a single mutated *BTK* gene results in XLA.
Since males have one X chromosome (XY), a single mutated *BTK* gene results in XLA.![]()
![]() Females have two X chromosomes (XX). If they inherit one mutated *BTK* gene, they are typically carriers but do not usually show symptoms because they have a functional copy on the other X chromosome. In rare cases, due to skewed X-inactivation, a carrier female might exhibit mild symptoms.
Females have two X chromosomes (XX). If they inherit one mutated *BTK* gene, they are typically carriers but do not usually show symptoms because they have a functional copy on the other X chromosome. In rare cases, due to skewed X-inactivation, a carrier female might exhibit mild symptoms.![]() Immunodeficiency:
Immunodeficiency:![]()
![]() The absence of mature B cells and antibodies (IgG, IgA, IgM, IgE) makes individuals highly susceptible to infections.
The absence of mature B cells and antibodies (IgG, IgA, IgM, IgE) makes individuals highly susceptible to infections.![]()
![]() Antibodies are vital for fighting off bacteria, viruses, and other pathogens. Without them, the body struggles to eliminate infections.
Antibodies are vital for fighting off bacteria, viruses, and other pathogens. Without them, the body struggles to eliminate infections.![]() Symptoms:
Symptoms:![]()
![]() Affected infants are usually protected for the first few months of life by antibodies acquired from their mothers during pregnancy.
Affected infants are usually protected for the first few months of life by antibodies acquired from their mothers during pregnancy.![]()
![]() Symptoms typically begin to appear between 6 months and 2 years of age as maternal antibodies wane.
Symptoms typically begin to appear between 6 months and 2 years of age as maternal antibodies wane.![]()
![]() Recurrent bacterial infections are the hallmark of XLA. Common infections include:
Recurrent bacterial infections are the hallmark of XLA. Common infections include:![]()
![]() Ear infections (otitis media)
Ear infections (otitis media)![]()
![]() Sinus infections (sinusitis)
Sinus infections (sinusitis)![]()
![]() Pneumonia
Pneumonia![]()
![]() Skin infections (pyoderma)
Skin infections (pyoderma)![]()
![]() Septicemia (blood poisoning)
Septicemia (blood poisoning)![]()
![]() Unusual susceptibility to certain viral infections, such as enteroviruses, which can lead to chronic neurological problems (e.g., meningoencephalitis).
Unusual susceptibility to certain viral infections, such as enteroviruses, which can lead to chronic neurological problems (e.g., meningoencephalitis).![]()
![]() Giardia lamblia infections in the intestines.
Giardia lamblia infections in the intestines.![]()
![]() Small tonsils and lymph nodes are often noted.
Small tonsils and lymph nodes are often noted.![]()
![]() Absence or reduced number of B cells in the blood.
Absence or reduced number of B cells in the blood.![]() Diagnosis:
Diagnosis:![]()
![]() Suspicion arises from recurrent infections, especially bacterial infections, in a young male child.
Suspicion arises from recurrent infections, especially bacterial infections, in a young male child.![]()
![]() Diagnosis is confirmed by:
Diagnosis is confirmed by:![]()
![]() Blood tests to measure immunoglobulin levels (IgG, IgA, IgM, IgE) - they will be very low or undetectable.
Blood tests to measure immunoglobulin levels (IgG, IgA, IgM, IgE) - they will be very low or undetectable.![]()
![]() Flow cytometry to count B cells in the blood - they will be virtually absent.
Flow cytometry to count B cells in the blood - they will be virtually absent.![]()
![]() Genetic testing to identify mutations in the *BTK* gene.
Genetic testing to identify mutations in the *BTK* gene.![]() Treatment:
Treatment:![]()
![]() The primary treatment is immunoglobulin replacement therapy (IgRT). This involves regular infusions or injections of antibodies from healthy donors to provide passive immunity.
The primary treatment is immunoglobulin replacement therapy (IgRT). This involves regular infusions or injections of antibodies from healthy donors to provide passive immunity.![]()
![]() IgRT helps prevent or reduce the severity and frequency of infections.
IgRT helps prevent or reduce the severity and frequency of infections.![]()
![]() Antibiotics are used to treat acute infections. Prophylactic antibiotics may be prescribed to prevent infections.
Antibiotics are used to treat acute infections. Prophylactic antibiotics may be prescribed to prevent infections.![]()
![]() Prompt treatment of infections is crucial to prevent complications.
Prompt treatment of infections is crucial to prevent complications.![]()
![]() Good hygiene practices are essential to minimize exposure to pathogens.
Good hygiene practices are essential to minimize exposure to pathogens.![]()
![]() Hematopoietic stem cell transplantation (HSCT) is sometimes considered, but is not generally considered a first-line treatment and carries significant risks.
Hematopoietic stem cell transplantation (HSCT) is sometimes considered, but is not generally considered a first-line treatment and carries significant risks.![]() Prognosis:
Prognosis:![]()
![]() With early diagnosis and consistent immunoglobulin replacement therapy, individuals with XLA can live relatively normal lives.
With early diagnosis and consistent immunoglobulin replacement therapy, individuals with XLA can live relatively normal lives.![]()
![]() However, they remain susceptible to infections and may develop chronic lung disease or other complications over time.
However, they remain susceptible to infections and may develop chronic lung disease or other complications over time.![]()
![]() Without treatment, XLA can be life-threatening due to severe and recurrent infections.
Without treatment, XLA can be life-threatening due to severe and recurrent infections.
In summary, XLA is a genetic disorder that leads to a lack of B cells and antibodies, causing severe immunodeficiency. Lifelong immunoglobulin replacement therapy is the cornerstone of treatment and significantly improves the prognosis.