Citrullinemia (Argininosuccinate Synthetase Deficiency, CTLN1)
Published: 18 Jun 2025
ICD9: 270.6 ICD10: E72.23 ICD11: 5C50.A3
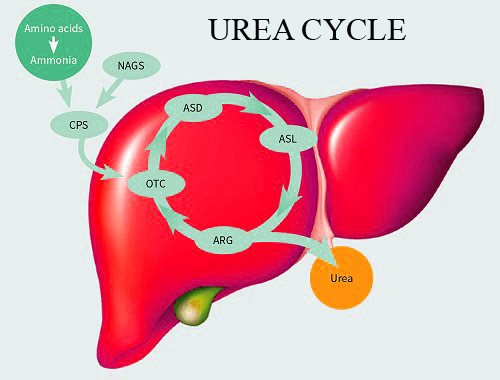
Citrullinemia type 1 (CTLN1), also known as Argininosuccinate Synthetase Deficiency (ASS1 Deficiency), is a rare, inherited metabolic disorder caused by a deficiency of the enzyme argininosuccinate synthetase (ASS1).
This enzyme plays a crucial role in the urea cycle, a pathway that removes ammonia, a toxic waste product, from the body.
Here's a breakdown:
![]() What it is: A genetic disorder affecting the urea cycle.
What it is: A genetic disorder affecting the urea cycle.![]() Cause: A deficiency in the argininosuccinate synthetase (ASS1) enzyme.
Cause: A deficiency in the argininosuccinate synthetase (ASS1) enzyme.![]() Effect: Impaired ammonia removal from the body, leading to a buildup of ammonia in the blood (hyperammonemia).
Effect: Impaired ammonia removal from the body, leading to a buildup of ammonia in the blood (hyperammonemia).![]() Inheritance: Autosomal recessive, meaning both parents must carry a copy of the mutated gene for the child to be affected.
Inheritance: Autosomal recessive, meaning both parents must carry a copy of the mutated gene for the child to be affected.
Key Aspects and Symptoms:![]() Hyperammonemia: This is the hallmark of the disorder. High levels of ammonia are toxic to the brain and other organs.
Hyperammonemia: This is the hallmark of the disorder. High levels of ammonia are toxic to the brain and other organs.![]() Types and Severity: CTLN1 can manifest in different forms:
Types and Severity: CTLN1 can manifest in different forms:![]()

![]() Neonatal-onset: The most severe form. Symptoms appear within the first few days of life.
Neonatal-onset: The most severe form. Symptoms appear within the first few days of life.![]()
![]() Symptoms: Lethargy, poor feeding, vomiting, seizures, coma, and potentially death. This form requires immediate medical intervention.
Symptoms: Lethargy, poor feeding, vomiting, seizures, coma, and potentially death. This form requires immediate medical intervention.![]()
![]() Late-onset: Symptoms may appear later in infancy, childhood, or even adulthood.
Late-onset: Symptoms may appear later in infancy, childhood, or even adulthood.![]()
![]() Symptoms: Can be triggered by illness, stress, or high-protein diets. May include: vomiting, confusion, seizures, ataxia (lack of coordination), behavioral changes, and developmental delays (in children). Sometimes it can even present as psychiatric symptoms.
Symptoms: Can be triggered by illness, stress, or high-protein diets. May include: vomiting, confusion, seizures, ataxia (lack of coordination), behavioral changes, and developmental delays (in children). Sometimes it can even present as psychiatric symptoms.![]() Diagnosis:
Diagnosis:![]()
![]() Newborn screening: In some regions, newborn screening programs test for citrullinemia.
Newborn screening: In some regions, newborn screening programs test for citrullinemia.![]()
![]() Blood tests: Elevated levels of ammonia and citrulline in the blood are indicative of the condition.
Blood tests: Elevated levels of ammonia and citrulline in the blood are indicative of the condition.![]()
![]() Genetic testing: Confirms the diagnosis by identifying mutations in the *ASS1* gene.
Genetic testing: Confirms the diagnosis by identifying mutations in the *ASS1* gene.![]()
![]() Urine tests: Measuring organic acids in the urine can help support the diagnosis.
Urine tests: Measuring organic acids in the urine can help support the diagnosis.![]() Treatment: The goal of treatment is to lower ammonia levels and prevent future episodes of hyperammonemia. Treatment strategies include:
Treatment: The goal of treatment is to lower ammonia levels and prevent future episodes of hyperammonemia. Treatment strategies include:![]()
![]() Dietary management: A low-protein diet to reduce ammonia production. Often supplemented with special formulas.
Dietary management: A low-protein diet to reduce ammonia production. Often supplemented with special formulas.![]()
![]() Medications:
Medications:![]()
![]() Ammonia scavengers: Medications like sodium phenylbutyrate or sodium benzoate help the body eliminate ammonia.
Ammonia scavengers: Medications like sodium phenylbutyrate or sodium benzoate help the body eliminate ammonia.![]()
![]() Arginine supplementation: Arginine is a precursor in the urea cycle and can help to enhance ammonia detoxification in some cases.
Arginine supplementation: Arginine is a precursor in the urea cycle and can help to enhance ammonia detoxification in some cases.![]()
![]() Hemodialysis or hemofiltration: May be necessary during acute hyperammonemic crises to rapidly remove ammonia from the blood.
Hemodialysis or hemofiltration: May be necessary during acute hyperammonemic crises to rapidly remove ammonia from the blood.![]()
![]() Liver transplantation: Can be a life-saving option in severe cases, as a healthy liver produces the necessary ASS1 enzyme.
Liver transplantation: Can be a life-saving option in severe cases, as a healthy liver produces the necessary ASS1 enzyme.![]() Prognosis: The prognosis varies depending on the severity of the disease and the effectiveness of treatment. Early diagnosis and prompt treatment are essential for improving outcomes. Neonatal-onset forms have a higher mortality rate. Lifelong management is typically required.
Prognosis: The prognosis varies depending on the severity of the disease and the effectiveness of treatment. Early diagnosis and prompt treatment are essential for improving outcomes. Neonatal-onset forms have a higher mortality rate. Lifelong management is typically required.
In summary: CTLN1 is a serious genetic disorder that requires careful management to prevent life-threatening complications. Understanding the condition and its treatment is critical for those affected and their families. If you suspect someone may have citrullinemia, it's important to seek medical attention immediately.