Cystic Fibrosis (Mucoviscidosis)
Published: 18 Jun 2025
ICD9: 277.01 ICD10: E84.11 ICD11: CA25
Cystic Fibrosis (CF), also known as mucoviscidosis, is a genetic disorder that primarily affects the lungs, but also the pancreas, liver, intestines, sinuses, and reproductive organs.
It's caused by a defect in the CFTR gene, which regulates the movement of salt and water in and out of cells.
Here's a breakdown of key aspects of Cystic Fibrosis:
1. The Root Cause: Faulty CFTR Gene
![]() Genetic Inheritance: CF is an autosomal recessive disorder. This means a person must inherit two copies of the mutated CFTR gene (one from each parent) to develop the disease. If they inherit only one copy, they are a carrier and usually show no symptoms, but can pass the gene to their children.
Genetic Inheritance: CF is an autosomal recessive disorder. This means a person must inherit two copies of the mutated CFTR gene (one from each parent) to develop the disease. If they inherit only one copy, they are a carrier and usually show no symptoms, but can pass the gene to their children.![]() CFTR Protein Malfunction: The CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) gene provides instructions for making a protein that acts as a channel, controlling the flow of chloride ions and water across cell membranes. In CF, the mutated CFTR gene results in a defective or missing protein.
CFTR Protein Malfunction: The CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) gene provides instructions for making a protein that acts as a channel, controlling the flow of chloride ions and water across cell membranes. In CF, the mutated CFTR gene results in a defective or missing protein.![]() Abnormal Mucus Production: The faulty CFTR protein disrupts the normal balance of salt and water, leading to the production of abnormally thick, sticky mucus.
Abnormal Mucus Production: The faulty CFTR protein disrupts the normal balance of salt and water, leading to the production of abnormally thick, sticky mucus.
2. Organs Affected and Symptoms![]() Lungs:
Lungs:![]()

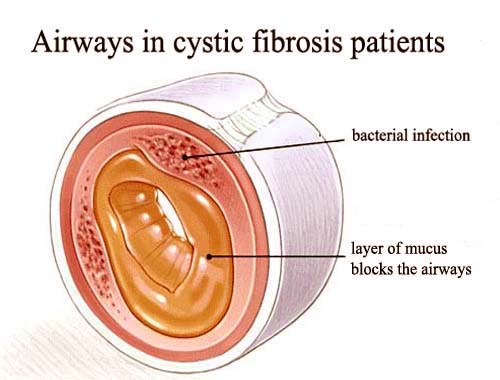
![]() Thick Mucus Buildup: Clogs airways, making it difficult to breathe.
Thick Mucus Buildup: Clogs airways, making it difficult to breathe.![]()
![]() Chronic Lung Infections: Thick mucus provides a breeding ground for bacteria, leading to frequent and persistent lung infections (e.g., pneumonia, bronchitis).
Chronic Lung Infections: Thick mucus provides a breeding ground for bacteria, leading to frequent and persistent lung infections (e.g., pneumonia, bronchitis).![]()
![]() Inflammation and Lung Damage: Chronic infections and inflammation lead to progressive lung damage, bronchiectasis (widening of the airways), and eventually respiratory failure.
Inflammation and Lung Damage: Chronic infections and inflammation lead to progressive lung damage, bronchiectasis (widening of the airways), and eventually respiratory failure.![]()
![]() Symptoms: Persistent cough (often with mucus), wheezing, shortness of breath, frequent lung infections, nasal congestion, sinus infections.
Symptoms: Persistent cough (often with mucus), wheezing, shortness of breath, frequent lung infections, nasal congestion, sinus infections.![]() Pancreas:
Pancreas:![]()
![]() Blocked Ducts: Thick mucus blocks the ducts that carry digestive enzymes from the pancreas to the small intestine.
Blocked Ducts: Thick mucus blocks the ducts that carry digestive enzymes from the pancreas to the small intestine.![]()
![]() Malabsorption: Without sufficient digestive enzymes, the body cannot properly absorb fats and proteins from food.
Malabsorption: Without sufficient digestive enzymes, the body cannot properly absorb fats and proteins from food.![]()
![]() Pancreatic Damage: Over time, the pancreas can be damaged, leading to diabetes.
Pancreatic Damage: Over time, the pancreas can be damaged, leading to diabetes.![]()
![]() Symptoms: Greasy, bulky stools, poor weight gain or growth, vitamin deficiencies.
Symptoms: Greasy, bulky stools, poor weight gain or growth, vitamin deficiencies.![]() Other Organs:
Other Organs:![]()
![]() Liver: Bile ducts can be blocked, leading to liver disease (e.g., cirrhosis).
Liver: Bile ducts can be blocked, leading to liver disease (e.g., cirrhosis).![]()
![]() Intestines: Meconium ileus (intestinal blockage in newborns), constipation.
Intestines: Meconium ileus (intestinal blockage in newborns), constipation.![]()
![]() Reproductive System: Most males with CF are infertile due to a blocked vas deferens (the tube that carries sperm). Women with CF may have reduced fertility due to thickened cervical mucus.
Reproductive System: Most males with CF are infertile due to a blocked vas deferens (the tube that carries sperm). Women with CF may have reduced fertility due to thickened cervical mucus.![]()
![]() Sweat Glands: People with CF have abnormally salty sweat. This is used in the sweat test, a diagnostic tool.
Sweat Glands: People with CF have abnormally salty sweat. This is used in the sweat test, a diagnostic tool.
3. Diagnosis![]() Newborn Screening: Many countries have newborn screening programs that test for CF using a blood test (immunoreactive trypsinogen or IRT test). A positive IRT test requires further testing.
Newborn Screening: Many countries have newborn screening programs that test for CF using a blood test (immunoreactive trypsinogen or IRT test). A positive IRT test requires further testing.![]() Sweat Test: Measures the amount of chloride in sweat. A high chloride level is a key indicator of CF.
Sweat Test: Measures the amount of chloride in sweat. A high chloride level is a key indicator of CF.![]() Genetic Testing: Identifies mutations in the CFTR gene.
Genetic Testing: Identifies mutations in the CFTR gene.![]() Clinical Evaluation: Doctors consider symptoms, family history, and results from other tests to make a diagnosis.
Clinical Evaluation: Doctors consider symptoms, family history, and results from other tests to make a diagnosis.
4. Treatment
There is no cure for CF, but treatments have significantly improved the quality of life and lifespan of people with CF. Treatment focuses on managing symptoms and preventing complications.![]() Airway Clearance Therapies: Techniques to loosen and remove mucus from the lungs (e.g., chest physiotherapy, high-frequency chest wall oscillation vests, breathing techniques).
Airway Clearance Therapies: Techniques to loosen and remove mucus from the lungs (e.g., chest physiotherapy, high-frequency chest wall oscillation vests, breathing techniques).![]() Medications:
Medications:![]()
![]() Bronchodilators: To open airways.
Bronchodilators: To open airways.![]()
![]() Mucolytics: To thin mucus.
Mucolytics: To thin mucus.![]()
![]() Antibiotics: To treat lung infections.
Antibiotics: To treat lung infections.![]()
![]() Anti-inflammatory drugs: To reduce inflammation in the lungs.
Anti-inflammatory drugs: To reduce inflammation in the lungs.![]()
![]() Pancreatic Enzyme Replacement Therapy: To aid digestion.
Pancreatic Enzyme Replacement Therapy: To aid digestion.![]()
![]() CFTR Modulators: These drugs target specific mutations in the CFTR gene and help the defective protein function better. They are not effective for all mutations, but have revolutionized treatment for many people with CF.
CFTR Modulators: These drugs target specific mutations in the CFTR gene and help the defective protein function better. They are not effective for all mutations, but have revolutionized treatment for many people with CF.![]() Nutritional Support: High-calorie diet, vitamin supplements (especially fat-soluble vitamins: A, D, E, K).
Nutritional Support: High-calorie diet, vitamin supplements (especially fat-soluble vitamins: A, D, E, K).![]() Lung Transplant: May be an option for people with severe lung disease.
Lung Transplant: May be an option for people with severe lung disease.![]() Exercise: Helps to clear mucus and improve lung function.
Exercise: Helps to clear mucus and improve lung function.
5. Prognosis
The prognosis for people with CF has improved dramatically over the past few decades due to advancements in treatment. The median survival age is now in the late 30s to 40s, and many people with CF are living longer, healthier lives. However, the disease is still serious, and life expectancy can vary depending on the severity of the disease and response to treatment.
Key Considerations:![]() Carrier Screening: Couples who are planning to have children can undergo carrier screening to determine their risk of having a child with CF.
Carrier Screening: Couples who are planning to have children can undergo carrier screening to determine their risk of having a child with CF.![]() Multidisciplinary Care: People with CF need comprehensive care from a team of healthcare professionals, including pulmonologists, gastroenterologists, dietitians, respiratory therapists, and social workers.
Multidisciplinary Care: People with CF need comprehensive care from a team of healthcare professionals, including pulmonologists, gastroenterologists, dietitians, respiratory therapists, and social workers.![]() Research: Ongoing research is focused on developing new and more effective treatments for CF, including gene therapy.
Research: Ongoing research is focused on developing new and more effective treatments for CF, including gene therapy.
In summary, Cystic Fibrosis is a complex genetic disorder that affects multiple organ systems, primarily the lungs and pancreas. The key feature is the production of thick, sticky mucus that leads to chronic infections and organ damage. While there is no cure, advances in treatment have significantly improved the lives of people with CF.