Langerhans cell histiocytosis (Hashimoto-Pritzger disease, Histiocytosis X)
Published: 18 Jun 2025
ICD9: 516.5 ICD10: J84.82 ICD11: 2B31.2Y
Langerhans cell histiocytosis (LCH), also known as Histiocytosis X and previously included Hashimoto-Pritzker disease (a self-healing form), is a rare disorder characterized by the abnormal proliferation and accumulation of specialized immune cells called Langerhans cells.
These cells are a type of dendritic cell normally found in the skin, lymph nodes, and other tissues, where they play a role in immune surveillance and antigen presentation.
Here's a breakdown of key aspects:
Key Features:
![]() Langerhans Cell Proliferation: The core feature is the excessive growth and accumulation of Langerhans cells.
Langerhans Cell Proliferation: The core feature is the excessive growth and accumulation of Langerhans cells.![]() Immune System Dysfunction: While the exact cause is unknown, LCH is believed to involve an immune system dysregulation, leading to the uncontrolled proliferation and migration of Langerhans cells.
Immune System Dysfunction: While the exact cause is unknown, LCH is believed to involve an immune system dysregulation, leading to the uncontrolled proliferation and migration of Langerhans cells.![]() Lesions and Tissue Damage: The accumulated Langerhans cells can form lesions in various organs and tissues, leading to localized damage and symptoms.
Lesions and Tissue Damage: The accumulated Langerhans cells can form lesions in various organs and tissues, leading to localized damage and symptoms.![]() Variable Presentation: LCH can range from a single, self-healing lesion to a severe, multi-system disease. The severity and the organs affected greatly influence the symptoms and prognosis.
Variable Presentation: LCH can range from a single, self-healing lesion to a severe, multi-system disease. The severity and the organs affected greatly influence the symptoms and prognosis.
Organs Affected:
LCH can affect virtually any organ system, but common sites include:![]() Bones: Bone lesions are frequent, often presenting as painful or swollen areas. Skull, femur, and vertebrae are commonly involved.
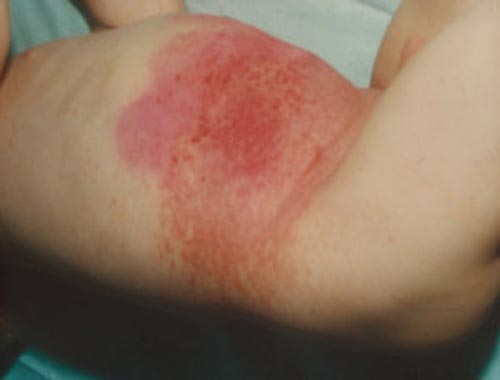
Bones: Bone lesions are frequent, often presenting as painful or swollen areas. Skull, femur, and vertebrae are commonly involved.![]() Skin: Skin lesions can manifest as rashes, papules, nodules, or ulcers.
Skin: Skin lesions can manifest as rashes, papules, nodules, or ulcers.![]() Lungs: Pulmonary LCH can cause shortness of breath, cough, and lung fibrosis. It's often associated with smoking.
Lungs: Pulmonary LCH can cause shortness of breath, cough, and lung fibrosis. It's often associated with smoking.![]() Pituitary Gland: Involvement of the pituitary gland can lead to diabetes insipidus (excessive thirst and urination).
Pituitary Gland: Involvement of the pituitary gland can lead to diabetes insipidus (excessive thirst and urination).![]() Liver, Spleen, Lymph Nodes: Involvement of these organs can lead to enlargement and dysfunction.
Liver, Spleen, Lymph Nodes: Involvement of these organs can lead to enlargement and dysfunction.![]() Central Nervous System (CNS): CNS involvement is less common but can cause neurological problems.
Central Nervous System (CNS): CNS involvement is less common but can cause neurological problems.![]() Hematopoietic System (Bone Marrow): Can cause cytopenias (low blood counts).
Hematopoietic System (Bone Marrow): Can cause cytopenias (low blood counts).
Classification and Severity:
LCH is classified based on the extent and distribution of the disease:![]() Single-System LCH (SS-LCH): Affects only one organ system (e.g., only bone or only skin). This generally has a better prognosis. Hashimoto-Pritzker disease, a rare congenital form, is often considered a self-healing type of SS-LCH.
Single-System LCH (SS-LCH): Affects only one organ system (e.g., only bone or only skin). This generally has a better prognosis. Hashimoto-Pritzker disease, a rare congenital form, is often considered a self-healing type of SS-LCH.![]() Multi-System LCH (MS-LCH): Affects multiple organ systems. This is generally more severe and requires more aggressive treatment. Multi-system disease can be further classified as:
Multi-System LCH (MS-LCH): Affects multiple organ systems. This is generally more severe and requires more aggressive treatment. Multi-system disease can be further classified as:![]()

![]() Risk-Organ Involvement: Involves organs like the liver, spleen, bone marrow, and/or lungs. This is associated with a poorer prognosis.
Risk-Organ Involvement: Involves organs like the liver, spleen, bone marrow, and/or lungs. This is associated with a poorer prognosis.![]()
![]() Non-Risk-Organ Involvement: Affects multiple organs but *not* the risk organs listed above.
Non-Risk-Organ Involvement: Affects multiple organs but *not* the risk organs listed above.
Symptoms:
The symptoms of LCH vary widely depending on the organs affected and the severity of the disease. Some common symptoms include:![]() Bone pain or swelling
Bone pain or swelling![]() Skin rash or lesions
Skin rash or lesions![]() Shortness of breath, cough
Shortness of breath, cough![]() Excessive thirst and urination (diabetes insipidus)
Excessive thirst and urination (diabetes insipidus)![]() Weight loss, fatigue
Weight loss, fatigue![]() Fever
Fever![]() Enlarged lymph nodes
Enlarged lymph nodes![]() Failure to thrive (in infants)
Failure to thrive (in infants)
Diagnosis:
Diagnosis typically involves:![]() Physical Examination and Medical History: To assess symptoms and risk factors.
Physical Examination and Medical History: To assess symptoms and risk factors.![]() Imaging Studies: X-rays, CT scans, MRI, and PET scans to identify lesions and assess organ involvement.
Imaging Studies: X-rays, CT scans, MRI, and PET scans to identify lesions and assess organ involvement.![]() Biopsy: A tissue sample is taken from a suspected lesion and examined under a microscope. Immunohistochemistry is performed to identify the characteristic CD207 (Langerin) and CD1a markers on Langerhans cells.
Biopsy: A tissue sample is taken from a suspected lesion and examined under a microscope. Immunohistochemistry is performed to identify the characteristic CD207 (Langerin) and CD1a markers on Langerhans cells.![]() Blood Tests: To assess organ function and identify any blood abnormalities.
Blood Tests: To assess organ function and identify any blood abnormalities.![]() Bone Marrow Aspiration and Biopsy: If bone marrow involvement is suspected.
Bone Marrow Aspiration and Biopsy: If bone marrow involvement is suspected.
Treatment:
Treatment depends on the extent and severity of the disease:![]() Observation: For single, localized lesions that may resolve spontaneously.
Observation: For single, localized lesions that may resolve spontaneously.![]() Local Therapy: Surgery, curettage, or topical corticosteroids for single bone or skin lesions.
Local Therapy: Surgery, curettage, or topical corticosteroids for single bone or skin lesions.![]() Systemic Chemotherapy: Used for multi-system LCH or single-system LCH that is not responsive to local therapy. Common chemotherapy agents include vinblastine, prednisone, methotrexate, and cladribine.
Systemic Chemotherapy: Used for multi-system LCH or single-system LCH that is not responsive to local therapy. Common chemotherapy agents include vinblastine, prednisone, methotrexate, and cladribine.![]() Targeted Therapy: In some cases, BRAF inhibitors (like vemurafenib) may be used if a BRAF V600E mutation is present in the Langerhans cells. This mutation is found in a significant proportion of LCH cases.
Targeted Therapy: In some cases, BRAF inhibitors (like vemurafenib) may be used if a BRAF V600E mutation is present in the Langerhans cells. This mutation is found in a significant proportion of LCH cases.![]() Stem Cell Transplantation: In severe, refractory cases.
Stem Cell Transplantation: In severe, refractory cases.![]() Supportive Care: Management of symptoms such as pain, diabetes insipidus, and other complications.
Supportive Care: Management of symptoms such as pain, diabetes insipidus, and other complications.
Prognosis:
The prognosis for LCH varies considerably depending on the extent of the disease, the organs involved, and the response to treatment.![]() Single-System LCH: Generally has a good prognosis with high survival rates.
Single-System LCH: Generally has a good prognosis with high survival rates.![]() Multi-System LCH: Prognosis is more variable and depends on the presence of risk-organ involvement and response to treatment. Risk-organ involvement is associated with a poorer prognosis. Relapse is also possible.
Multi-System LCH: Prognosis is more variable and depends on the presence of risk-organ involvement and response to treatment. Risk-organ involvement is associated with a poorer prognosis. Relapse is also possible.
Important Considerations:![]() Rare Disease: LCH is a rare disease, and diagnosis can be challenging.
Rare Disease: LCH is a rare disease, and diagnosis can be challenging.![]() Multidisciplinary Approach: Management of LCH often requires a multidisciplinary team of specialists, including oncologists, hematologists, dermatologists, endocrinologists, pulmonologists, and neurologists.
Multidisciplinary Approach: Management of LCH often requires a multidisciplinary team of specialists, including oncologists, hematologists, dermatologists, endocrinologists, pulmonologists, and neurologists.![]() Research: Ongoing research is aimed at better understanding the causes of LCH and developing more effective treatments.
Research: Ongoing research is aimed at better understanding the causes of LCH and developing more effective treatments.
In summary, Langerhans cell histiocytosis is a complex and variable disorder characterized by the abnormal proliferation of Langerhans cells. The diagnosis and management require careful evaluation and a tailored approach based on the individual patient's presentation and disease severity.