Nonketotic Hyperglycinemia (Glycine encephalopathy)
Published: 18 Jun 2025
ICD9: 270.7 ICD10: E72.51 ICD11: 5C50.70
Nonketotic Hyperglycinemia (NKH), also known as Glycine Encephalopathy, is a rare, inherited metabolic disorder that affects the brain and nervous system.
It's characterized by an accumulation of glycine, an amino acid, in the body's tissues and fluids, especially the brain. This build-up is toxic to the brain, causing a range of neurological problems.
Here's a breakdown of the key aspects:
![]() Cause: The most common cause is a deficiency in the glycine cleavage system (GCS), a complex of enzymes responsible for breaking down glycine. This deficiency is usually due to mutations in genes that encode for the proteins of the GCS. Less common causes can include defects in glycine receptors.
Cause: The most common cause is a deficiency in the glycine cleavage system (GCS), a complex of enzymes responsible for breaking down glycine. This deficiency is usually due to mutations in genes that encode for the proteins of the GCS. Less common causes can include defects in glycine receptors.![]() Mechanism: The exact mechanism by which high levels of glycine damage the brain isn't fully understood, but it's thought that glycine acts as an overstimulatory neurotransmitter (agonist) at certain receptors (especially NMDA receptors), leading to excitotoxicity (overstimulation of nerve cells to the point of damage or death).
Mechanism: The exact mechanism by which high levels of glycine damage the brain isn't fully understood, but it's thought that glycine acts as an overstimulatory neurotransmitter (agonist) at certain receptors (especially NMDA receptors), leading to excitotoxicity (overstimulation of nerve cells to the point of damage or death).![]() Symptoms: Symptoms vary widely in severity, ranging from mild to severe. There are different forms of NKH:
Symptoms: Symptoms vary widely in severity, ranging from mild to severe. There are different forms of NKH:![]()

![]() Neonatal/Severe NKH (most common): This form presents within the first few days of life with symptoms like:
Neonatal/Severe NKH (most common): This form presents within the first few days of life with symptoms like:![]()
![]() Lethargy and poor feeding
Lethargy and poor feeding![]()
![]() Hypotonia (floppiness)
Hypotonia (floppiness)![]()
![]() Seizures (often intractable and myoclonic)
Seizures (often intractable and myoclonic)![]()
![]() Apnea (episodes of stopped breathing)
Apnea (episodes of stopped breathing)![]()
![]() Developmental delay (severe)
Developmental delay (severe)![]()
![]() Intellectual disability (severe)
Intellectual disability (severe)![]()
![]() Late-Onset NKH: Symptoms appear later in infancy or childhood and may include:
Late-Onset NKH: Symptoms appear later in infancy or childhood and may include:![]()
![]() Intellectual disability (variable)
Intellectual disability (variable)![]()
![]() Seizures (variable)
Seizures (variable)![]()
![]() Spasticity (muscle stiffness)
Spasticity (muscle stiffness)![]()
![]() Movement disorders (e.g., chorea, dystonia)
Movement disorders (e.g., chorea, dystonia)![]()
![]() Behavioral problems
Behavioral problems![]()
![]() Attenuated NKH: This is a milder form with milder neurological symptoms that may not be apparent until later in life. Some individuals may have normal intelligence.
Attenuated NKH: This is a milder form with milder neurological symptoms that may not be apparent until later in life. Some individuals may have normal intelligence.![]() Diagnosis: Diagnosis is typically based on:
Diagnosis: Diagnosis is typically based on:![]()
![]() Elevated glycine levels in cerebrospinal fluid (CSF) compared to blood. The CSF/plasma glycine ratio is particularly important.
Elevated glycine levels in cerebrospinal fluid (CSF) compared to blood. The CSF/plasma glycine ratio is particularly important.![]()
![]() Genetic testing: Identifies mutations in genes associated with NKH (e.g., GLDC, AMT, GCSH).
Genetic testing: Identifies mutations in genes associated with NKH (e.g., GLDC, AMT, GCSH).![]()
![]() Clinical presentation: Symptoms and age of onset.
Clinical presentation: Symptoms and age of onset.![]()
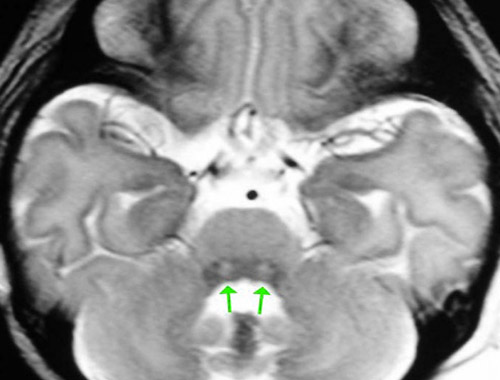
![]() Brain imaging (MRI): May show characteristic patterns of brain damage.
Brain imaging (MRI): May show characteristic patterns of brain damage.![]() Treatment: There is no cure for NKH. Treatment is focused on managing symptoms and reducing glycine levels:
Treatment: There is no cure for NKH. Treatment is focused on managing symptoms and reducing glycine levels:![]()
![]() Sodium benzoate: Helps to remove glycine from the body.
Sodium benzoate: Helps to remove glycine from the body.![]()
![]() Dextromethorphan (DXM): An NMDA receptor antagonist that may help reduce glycine's excitotoxic effects.
Dextromethorphan (DXM): An NMDA receptor antagonist that may help reduce glycine's excitotoxic effects.![]()
![]() Strychnine: (Rarely used, highly controversial) A glycine receptor antagonist used in some severe cases to block the effects of glycine.
Strychnine: (Rarely used, highly controversial) A glycine receptor antagonist used in some severe cases to block the effects of glycine.![]()
![]() Antiepileptic medications: To control seizures.
Antiepileptic medications: To control seizures.![]()
![]() Supportive care: Physical therapy, occupational therapy, speech therapy, and nutritional support.
Supportive care: Physical therapy, occupational therapy, speech therapy, and nutritional support.![]() Prognosis: The prognosis for individuals with severe NKH is generally poor. Many affected infants do not survive beyond infancy or early childhood. Those with later-onset or attenuated forms may have a better prognosis, depending on the severity of their symptoms.
Prognosis: The prognosis for individuals with severe NKH is generally poor. Many affected infants do not survive beyond infancy or early childhood. Those with later-onset or attenuated forms may have a better prognosis, depending on the severity of their symptoms.![]() Inheritance: NKH is an autosomal recessive disorder. This means that both parents must carry a copy of the mutated gene for their child to be affected. If both parents are carriers, there is a 25% chance with each pregnancy that their child will inherit the condition, a 50% chance that their child will be a carrier, and a 25% chance that their child will be unaffected.
Inheritance: NKH is an autosomal recessive disorder. This means that both parents must carry a copy of the mutated gene for their child to be affected. If both parents are carriers, there is a 25% chance with each pregnancy that their child will inherit the condition, a 50% chance that their child will be a carrier, and a 25% chance that their child will be unaffected.
In summary, Nonketotic Hyperglycinemia (Glycine Encephalopathy) is a serious metabolic disorder characterized by a build-up of glycine in the brain, leading to significant neurological problems. Early diagnosis and treatment are crucial to managing the condition and improving outcomes.