Urea cycle disorder
Published: 18 Jun 2025
ICD9: 270.6 ICD10: E72.20 ICD11: 5C50.AZ
A urea cycle disorder (UCD) is a genetic condition that causes ammonia to build up in the blood.
Here's a breakdown:
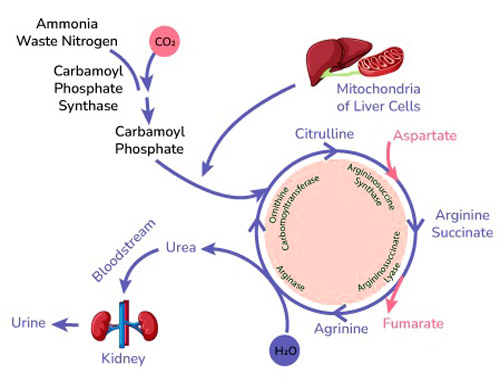
What is the Urea Cycle?
![]() The urea cycle is a series of biochemical reactions that occur in the liver.
The urea cycle is a series of biochemical reactions that occur in the liver.![]() Its primary purpose is to remove ammonia, a toxic waste product of protein metabolism, from the body.
Its primary purpose is to remove ammonia, a toxic waste product of protein metabolism, from the body.![]() Ammonia is converted into urea, a less toxic substance that can be excreted in urine.
Ammonia is converted into urea, a less toxic substance that can be excreted in urine.
What Happens in a UCD?![]() In UCDs, one of the enzymes in the urea cycle is either deficient or completely missing.
In UCDs, one of the enzymes in the urea cycle is either deficient or completely missing.![]() This deficiency prevents the cycle from functioning correctly, leading to a buildup of ammonia in the blood (hyperammonemia).
This deficiency prevents the cycle from functioning correctly, leading to a buildup of ammonia in the blood (hyperammonemia).![]() Ammonia is toxic, especially to the brain, and can cause irreversible brain damage, coma, and even death if not treated promptly.
Ammonia is toxic, especially to the brain, and can cause irreversible brain damage, coma, and even death if not treated promptly.
Causes![]() UCDs are caused by genetic mutations (changes) in genes that provide instructions for making the enzymes of the urea cycle.
UCDs are caused by genetic mutations (changes) in genes that provide instructions for making the enzymes of the urea cycle.![]() Most UCDs are inherited in an autosomal recessive pattern, meaning both parents must carry a copy of the mutated gene for the child to be affected. There is also one X-linked urea cycle disorder.
Most UCDs are inherited in an autosomal recessive pattern, meaning both parents must carry a copy of the mutated gene for the child to be affected. There is also one X-linked urea cycle disorder.
Types of UCDs
There are several different types of UCDs, depending on which enzyme is affected:![]() Ornithine transcarbamylase deficiency (OTC): Most common UCD. X-linked.
Ornithine transcarbamylase deficiency (OTC): Most common UCD. X-linked.![]() Carbamoyl phosphate synthetase I deficiency (CPS1):
Carbamoyl phosphate synthetase I deficiency (CPS1):![]() Argininosuccinate synthetase deficiency (ASS1), also known as citrullinemia type I:
Argininosuccinate synthetase deficiency (ASS1), also known as citrullinemia type I:![]() Argininosuccinate lyase deficiency (ASL):
Argininosuccinate lyase deficiency (ASL):![]() Arginase deficiency (ARG1):
Arginase deficiency (ARG1):![]() N-acetylglutamate synthase deficiency (NAGS): Rarest UCD.
N-acetylglutamate synthase deficiency (NAGS): Rarest UCD.
Symptoms
Symptoms vary depending on the severity of the deficiency and the age of onset, but may include:![]() In newborns:
In newborns:![]()

![]() Lethargy
Lethargy![]()
![]() Poor feeding
Poor feeding![]()
![]() Vomiting
Vomiting![]()
![]() Irritability
Irritability![]()
![]() Rapid breathing
Rapid breathing![]()
![]() Seizures
Seizures![]()
![]() Coma
Coma![]() In older children and adults:
In older children and adults:![]()
![]() Confusion
Confusion![]()
![]() Headaches
Headaches![]()
![]() Behavioral changes
Behavioral changes![]()
![]() Slurred speech
Slurred speech![]()
![]() Ataxia (lack of coordination)
Ataxia (lack of coordination)![]()
![]() Avoidance of high-protein foods
Avoidance of high-protein foods![]()
![]() Cyclic vomiting
Cyclic vomiting![]()
![]() Seizures
Seizures![]()
![]() Coma
Coma
Diagnosis![]() Blood tests: Elevated ammonia levels are a key indicator. Other tests to check amino acid levels (citrulline, arginine, etc.) are important.
Blood tests: Elevated ammonia levels are a key indicator. Other tests to check amino acid levels (citrulline, arginine, etc.) are important.![]() Urine tests:
Urine tests:![]() Genetic testing: To identify the specific gene mutation.
Genetic testing: To identify the specific gene mutation.![]() Enzyme assays: (Less commonly done now with readily available genetic testing) Measure the activity of the urea cycle enzymes.
Enzyme assays: (Less commonly done now with readily available genetic testing) Measure the activity of the urea cycle enzymes.
Treatment
Treatment focuses on lowering ammonia levels and preventing future episodes of hyperammonemia. It is a lifelong undertaking. Treatment options include:![]() Dietary management:
Dietary management:![]()
![]() Restricting protein intake.
Restricting protein intake.![]()
![]() Providing special formulas and foods low in protein but high in calories.
Providing special formulas and foods low in protein but high in calories.![]() Medications:
Medications:![]()
![]() Ammonia scavengers: Drugs like sodium benzoate and sodium phenylbutyrate help the body get rid of ammonia through alternative pathways.
Ammonia scavengers: Drugs like sodium benzoate and sodium phenylbutyrate help the body get rid of ammonia through alternative pathways.![]()
![]() Arginine or citrulline supplementation: Some UCDs benefit from supplementation.
Arginine or citrulline supplementation: Some UCDs benefit from supplementation.![]() Hemodialysis or hemofiltration: In severe cases, these procedures can rapidly remove ammonia from the blood.
Hemodialysis or hemofiltration: In severe cases, these procedures can rapidly remove ammonia from the blood.![]() Liver transplantation: A liver transplant can cure UCDs by providing a functional urea cycle. This is considered a definitive treatment for some, but carries its own risks and requires lifelong immunosuppression.
Liver transplantation: A liver transplant can cure UCDs by providing a functional urea cycle. This is considered a definitive treatment for some, but carries its own risks and requires lifelong immunosuppression.
Prognosis
The prognosis for individuals with UCDs depends on the severity of the deficiency, the age of diagnosis, and how well the condition is managed. Early diagnosis and treatment are crucial to minimize brain damage and improve outcomes. Lifelong adherence to treatment is necessary.
Importance of Newborn Screening
Many states include UCDs in their newborn screening programs, which allows for early diagnosis and intervention, significantly improving outcomes. This is typically done through a blood test (heel prick) shortly after birth.
It is important to consult with a medical professional for accurate diagnosis and treatment of Urea Cycle Disorders. This is a complex condition, and management should be individualized.