Wilson's disease
Published: 18 Jun 2025
ICD9: 275.1 ICD10: E83.01 ICD11: 5C64.00
Wilson's disease is a rare, inherited disorder that prevents the body from properly removing excess copper.
This leads to a buildup of copper in the liver, brain, and other vital organs. This build-up can cause serious health problems, including liver damage, neurological problems, and psychiatric issues.
Here's a more detailed breakdown:
Key Aspects of Wilson's Disease:
![]() Genetic Cause: It's caused by a mutation in the *ATP7B* gene. This gene provides instructions for making a protein that helps the liver excrete copper into bile (a digestive fluid). When this gene is mutated, the protein doesn't function correctly, and copper accumulates. It's an autosomal recessive disorder, meaning a person must inherit two copies of the mutated gene (one from each parent) to develop the disease. If someone inherits only one copy, they are a carrier but usually don't show symptoms.
Genetic Cause: It's caused by a mutation in the *ATP7B* gene. This gene provides instructions for making a protein that helps the liver excrete copper into bile (a digestive fluid). When this gene is mutated, the protein doesn't function correctly, and copper accumulates. It's an autosomal recessive disorder, meaning a person must inherit two copies of the mutated gene (one from each parent) to develop the disease. If someone inherits only one copy, they are a carrier but usually don't show symptoms.![]() Copper Accumulation: Normally, copper is absorbed from food in the small intestine. The liver processes the copper, using some of it and excreting the excess into bile for elimination from the body. In Wilson's disease, the liver is unable to excrete copper properly. This leads to copper accumulating in the liver, eventually causing damage. The copper then spills over into the bloodstream and deposits in other organs, primarily the brain, kidneys, and corneas.
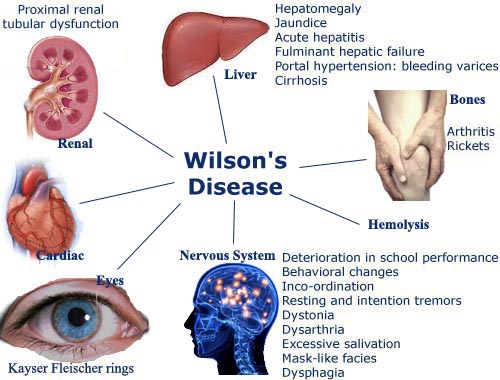
Copper Accumulation: Normally, copper is absorbed from food in the small intestine. The liver processes the copper, using some of it and excreting the excess into bile for elimination from the body. In Wilson's disease, the liver is unable to excrete copper properly. This leads to copper accumulating in the liver, eventually causing damage. The copper then spills over into the bloodstream and deposits in other organs, primarily the brain, kidneys, and corneas.![]() Symptoms: Symptoms vary widely among individuals and depend on which organs are affected and the amount of copper accumulation. They can develop at any age, but often appear between the ages of 5 and 35. Common symptoms include:
Symptoms: Symptoms vary widely among individuals and depend on which organs are affected and the amount of copper accumulation. They can develop at any age, but often appear between the ages of 5 and 35. Common symptoms include:![]()

![]() Liver Problems:
Liver Problems:![]()
![]() Fatigue, weakness
Fatigue, weakness![]()
![]() Abdominal pain
Abdominal pain![]()
![]() Jaundice (yellowing of the skin and eyes)
Jaundice (yellowing of the skin and eyes)![]()
![]() Swelling in the legs and abdomen
Swelling in the legs and abdomen![]()
![]() Liver failure
Liver failure![]()
![]() Neurological Problems:
Neurological Problems:![]()
![]() Tremors
Tremors![]()
![]() Difficulty with coordination
Difficulty with coordination![]()
![]() Slurred speech
Slurred speech![]()
![]() Muscle stiffness
Muscle stiffness![]()
![]() Difficulty swallowing
Difficulty swallowing![]()
![]() Psychiatric Problems:
Psychiatric Problems:![]()
![]() Depression
Depression![]()
![]() Anxiety
Anxiety![]()
![]() Personality changes
Personality changes![]()
![]() Psychosis
Psychosis![]()
![]() Kayser-Fleischer Rings: These are brownish rings around the cornea of the eye, caused by copper deposits. They are a characteristic sign of Wilson's disease, but are not always present, especially in early stages.
Kayser-Fleischer Rings: These are brownish rings around the cornea of the eye, caused by copper deposits. They are a characteristic sign of Wilson's disease, but are not always present, especially in early stages.![]()
![]() Other Symptoms: Kidney problems, anemia, joint pain.
Other Symptoms: Kidney problems, anemia, joint pain.![]() Diagnosis: Diagnosis typically involves a combination of:
Diagnosis: Diagnosis typically involves a combination of:![]()
![]() Physical Exam and Medical History: Evaluating symptoms and family history.
Physical Exam and Medical History: Evaluating symptoms and family history.![]()
![]() Eye Exam: Checking for Kayser-Fleischer rings.
Eye Exam: Checking for Kayser-Fleischer rings.![]()
![]() Blood Tests: Measuring copper levels in the blood, ceruloplasmin levels (a copper-carrying protein), and liver function tests. Low ceruloplasmin is typical.
Blood Tests: Measuring copper levels in the blood, ceruloplasmin levels (a copper-carrying protein), and liver function tests. Low ceruloplasmin is typical.![]()
![]() Urine Tests: Measuring copper levels in the urine.
Urine Tests: Measuring copper levels in the urine.![]()
![]() Liver Biopsy: Taking a small sample of liver tissue to measure copper content and assess liver damage.
Liver Biopsy: Taking a small sample of liver tissue to measure copper content and assess liver damage.![]()
![]() Genetic Testing: Analyzing the *ATP7B* gene for mutations.
Genetic Testing: Analyzing the *ATP7B* gene for mutations.![]() Treatment: Wilson's disease is treatable, and early diagnosis and treatment are crucial to prevent serious complications. Treatment focuses on removing excess copper from the body and preventing its reaccumulation. Treatment options include:
Treatment: Wilson's disease is treatable, and early diagnosis and treatment are crucial to prevent serious complications. Treatment focuses on removing excess copper from the body and preventing its reaccumulation. Treatment options include:![]()
![]() Chelating Agents: These medications bind to copper, allowing it to be excreted in the urine. Common chelating agents include penicillamine and trientine.
Chelating Agents: These medications bind to copper, allowing it to be excreted in the urine. Common chelating agents include penicillamine and trientine.![]()
![]() Zinc Acetate: Zinc blocks the absorption of copper from food in the digestive tract.
Zinc Acetate: Zinc blocks the absorption of copper from food in the digestive tract.![]()
![]() Liver Transplant: In severe cases of liver failure, a liver transplant may be necessary.
Liver Transplant: In severe cases of liver failure, a liver transplant may be necessary.![]()
![]() Dietary Changes: Limiting copper intake by avoiding foods high in copper, such as liver, shellfish, chocolate, nuts, and mushrooms.
Dietary Changes: Limiting copper intake by avoiding foods high in copper, such as liver, shellfish, chocolate, nuts, and mushrooms.![]() Importance of Early Diagnosis: If left untreated, Wilson's disease can lead to severe liver damage, neurological damage, and even death. Early diagnosis and treatment can significantly improve the prognosis and quality of life for individuals with this condition. Lifelong treatment is usually required.
Importance of Early Diagnosis: If left untreated, Wilson's disease can lead to severe liver damage, neurological damage, and even death. Early diagnosis and treatment can significantly improve the prognosis and quality of life for individuals with this condition. Lifelong treatment is usually required.
In summary, Wilson's disease is a serious genetic disorder that can have devastating consequences if left untreated. However, with early diagnosis and proper management, individuals with Wilson's disease can live relatively normal lives.